CRISPR-Quest Gene Editing Test
Screening Mutant Clones after CRISPR Gene Editing
The CRISPR-Quest gene editing test uses XNA technology to screen for the mutants created by CRISPR/CAS 9 gene editing technique. The wildtype gene sequence that covers the expected mutation site is chosen for the custom XNA design. The custom-synthesized XNA then is used in a qPCR reaction to block the wildtype sequence amplification but it selectively amplifies the DNA containing the mutant sequence. The screening method can be used to screen for mutants from selected pools, or screen/confirm individual clones that contain the desired mutations. The mutant DNA can be further sequenced by Sanger method. The method is especially effective for high-throughput screening of guide RNAs and mutant clones.
The example below shows the XNA design for the WTAP gene. The XNA sequence used for the design, the PAM sequence and cleavage site are shown below.
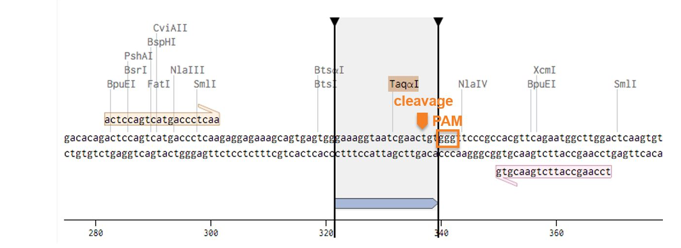
We would need the information to help you design the primers and XNA for screening the mutants and further sequencing the mutant using Sanger sequencing. Contact us for XNA design used for gene editing mutant screening.
GET A QUOTE NOW
How Does the XNA Screening Work?
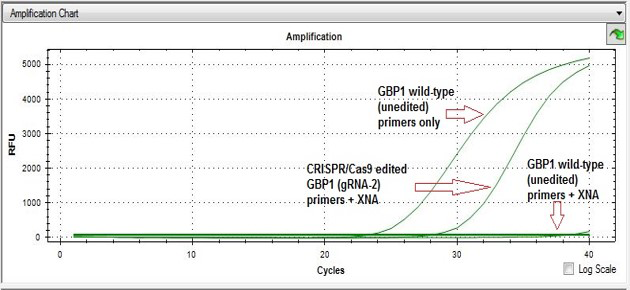
In a typical Non-homology End Joining (NHEJ) mutant screening process after the CRISPR gene-editing event, one can run a SYBR Green qPCR using a positive control (wild-type DNA plus primers, no XNA), a negative control (wildtype DNA plus primers, with XNA), and the sample DNA (from cell pool or individual clones). The amplification plot for a mutant due to a nucleotide substitution, or an indel, will be located between the plots for positive control and the negative control, as shown on the right. If there is no mutation present, the sample amplification plot will be similar to the negative control plot.
Based on the Cq value difference (∆Cq) between the Cq for the mutant and the positive control, the percentage of the edited cell proportion can be roughly estimated, although it is not accurate.
Editing Events %=2 –∆Cq x100%
For instance, if the Cq for the positive control is 25 cycles and the Cq for the mutant is 27 cycles, the ∆Cq is 2 and the editing event is 25%. However, for mutant screening or gRNA screening, direct comparison of Cq value of each sample with that of the wildtype should give clues as to which gRNA works better out of the 3 or 4 gRNAs used.
We can synthesize the XNA enough for 100 and 500 reactions for your screening needs. The reactions come with the qPCR mix, one pair of qPCR primers, XNA, positive and negative controls. Larger batch of custom synthesis is also available.
CRISPR Makes it Easier for Creating Stable Cell Lines with Particular Gene Mutations
Genome editing by the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) system has revolutionized the creation of gene mutations. The research community’s rapid acceptance of the CRISPR/Cas9 technology has made gene editing a routine practice for molecular biology labs. Using CRISPR, creating gene knockout mutations is remarkably easy to do for any gene of interest. No more traditional gene knockout and siRNA or shRNA knock-down techniques are necessary.
Challenges for Screening for Desired Mutants from CRISPR Gene Editing
One of the biggest challenges in using the CRISPR system is the slow turnaround time from the introduction of the CRISPR system to identification of the clones with desired mutations. This may be due to the low efficiency of the current technology and the nature of the gene disruption mechanism.
After transfection with the guide RNA (gRNA) and Cas9 system, the cells or cell clones with possible mutations are selected or enriched either with antibiotics selection or with reporter gene expression sorting. The real challenge is to select the low percentage of clones that contain the mutants of interest from the large proportion of the clones with wild-type sequences.
Most of the methods used to screen for clones with desired gene mutations do not work efficiently and/or take much longer time to design and screen. Current methods include:
PCR followed by restriction enzyme digestions
PCR followed by mismatch assays
High resolution melting analysis
Capillary electrophoresis
Sanger sequencing combined with PCR
Western blot
Competition-based PCR method
Utilizing XNA Technology for Mutant Screening
XNA is the Optimal Choice for Screening Mutant Clones after CRISPR Gene Editing Compared to Other Technologies.
Regular PCR methods fail to identify the mutant in majority of the wildtype background because the gene editing event occurs in <1% frequency. Our XNA technology can selectively detect the mutants created by Cas9 gene editing system by suppressing the wildtype sequence at the expected mutation site, regardless of the fact that the mutation is a point mutation or Indel mutation. In a 3-hour qPCR reaction, the mutant DNA can be identified if present in the pool DNA.
In addition to identifying the mutant clones, the technique also enables empirical validation of guide RNA efficiency and measurement of the ratio of HDR: NHEJ at a targeted locus.
Fast Results
Just set up the qPCR reaction and get results in 3 hours. In contrast, other methods are tedious and need much more hands-on time.
No Manual Design
We will design the XNA for you if you provide your wild-type PAM flanking sequence. Order the XNA and qPCR reactions when gRNA is ordered.
Ultra-Sensitivity
Cell lines with low transfection efficiency or poor editing efficiency because of gRNA design often generate low percentage gene editing and need more clones to screen. XNA technology helps in identifying
CRISPR-Quest Gene Editing Test Supporting Data
qPCR Results
The rare cell with a single-base change is isolated using XNA technology as a measurement tool, then enriched sequentially until a pure clone is obtained.
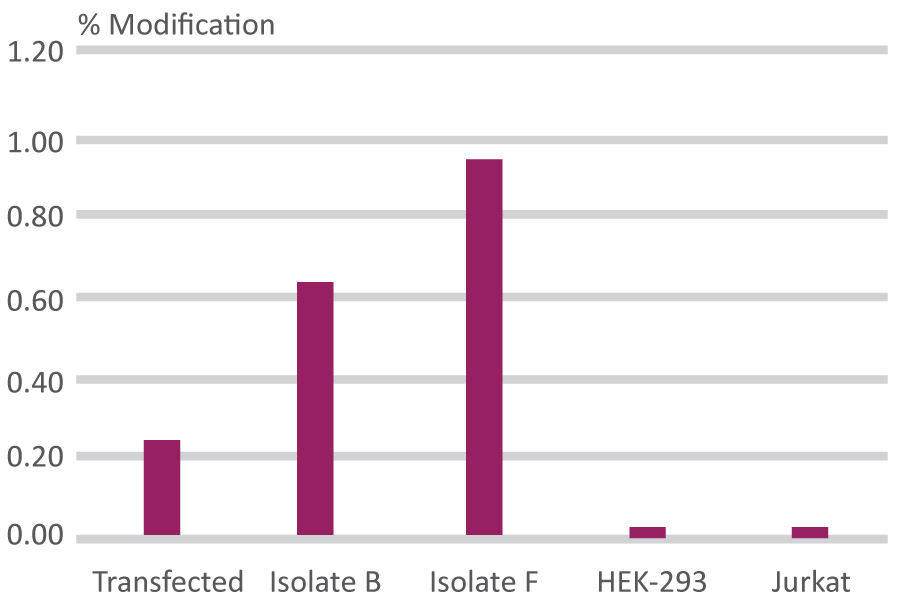
Calculated Percentage Modification
Compares
| CRISPR-Quest BioRad/Taq | CRISPR-Quest NEB | Amplicon Sequencing | T7 Endonucleases | |
|---|---|---|---|---|
| Transfected Pool | 21% | 15% | 26.5% | 20% |
| Isolate B | 62% | 50% | - | - |
| Isolate F | 99% | 95% | 92.6% | 7.5% |
| HEK-295 | 0% | 0% | - | - |
| Jurkat | -0% | -0% | - | - |
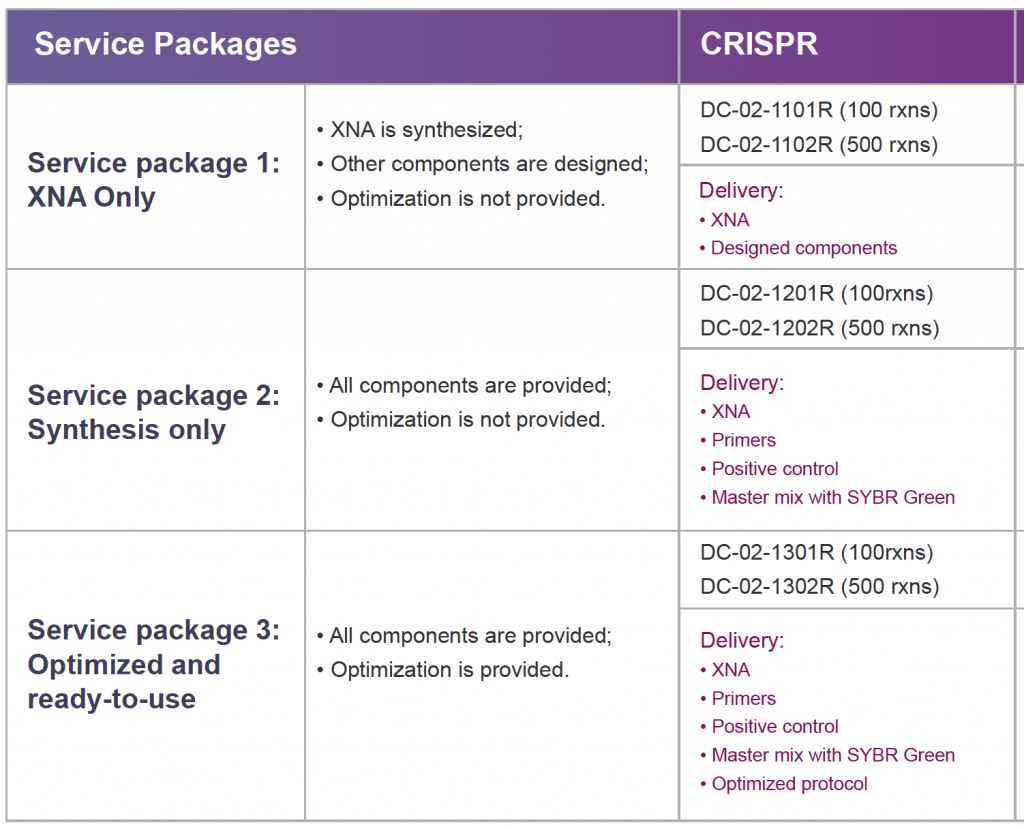
CRISPR-Quest Gene Editing Test Product Specifications
Product Catalog Number
Intended Use
For research use only
Pack Size
100 Reactions and 500 Reactions
Stability
Stable for 12 Months at -25°C to -15°C